Information for professionals about the 22q11 Deletion Syndrome Multi-Disciplinary Team Clinic
This clinic is for children and young people who have 22q11 deletion syndrome and their families. The role of the clinic is to bring together affected children, families with a specialist multi-disciplinary team to help coordinate and guide care of these patients across Wales. For effective care, local community services are essential, and early involvement of a community paediatrician is strongly encouraged. The following content is intended as an aid to healthcare workers involved in the care of patients with this common condition, and gratefully acknowledges the Max Appeal consensus document from clinicians’ relevant summaries on 22q11 deletion syndrome as a key source.
Aims of the multi-disciplinary clinic
- Children and adolescents with 22q11 deletion are all very different. Some show a wide range of problems while others hardly at all. As a multi-disciplinary team we want to ensure that all the appropriate assessments have been undertaken.
- The clinic is for children and young people who have a diagnosis of 22q11 Deletion Syndrome and their families.
- All newly diagnosed patients will be offered an appointment on the next available clinic
- The clinics are held at the UHW 3 times per year.
- Families can request to attend the clinic at any point during their treatment, particularly if there are concerns.
- Specialities present include: Clinical Immunology, Cleft and Maxillofacial Surgery, Speech and Language Therapy, Clinical Psychology, Clinical Genetics, and a Family Support Representative of the Max Appeal.
- In depth diagnosis or treatment will not be provided at the clinic. Where necessary this will be provided at a future appointment with the relevant professional.
- The purpose of the clinic is to check that all important assessments have been arranged or completed.
- Following the clinic, a letter will be sent to the GP outlining the outcome of the clinic and copied to all relevant professionals.
Referral form:
When referring a patient to the 22q11 deletion syndrome MDT clinic, please consider including the information in the following document:
Prompts for 22q11 MDT referral
Background Information
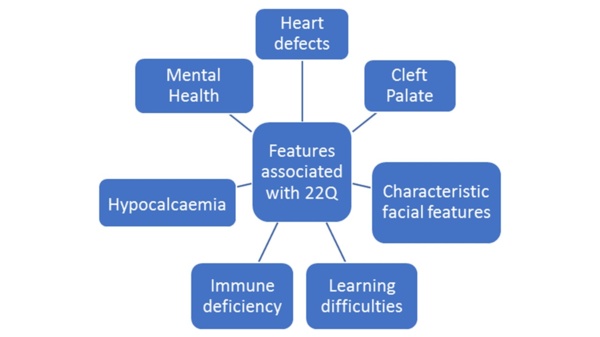
In 1965, ‘DiGeorge Syndrome’ was first described as a triad of cardiac defects, hypocalcaemia and immunodeficiency (1, 2). In the 1990s, fluorescence in situ hybridization identified a deletion at band 11.2 on the long arm of chromosome 22 as the cause of this and overlapping syndromes, such as velo-cardio-facial syndrome, DiGeorge Syndrome and Shprintzen syndrome (3). Now it is recognised the features are due to the same deletion, the syndrome is classified as the “22q11.2 deletion syndrome”.
Array comparative genomic hybridisation is now widely used in children with learning disabilities, providing more accurate information on 22q deletions and duplications. Children with subtle features are increasingly being tested, possibly leading to more 22q11 deletion syndrome diagnoses (4, 5, 6). It is the commonest deletion syndrome, with incidence estimates range from 1/1000 - 1/6000 births, with the most common being 1/4000 (1-4, 7-8).
Inheritance follows an autosomal dominant fashion. However, approximately 90% of cases are spontaneous mutations (1, 3). It is recommended that parental chromosomes are checked as they may have undiagnosed 22q11.2 deletion syndrome, being asymptomatic or having mild features. Parents may wish to use prenatal testing on future pregnancies (1, 5, 9). The heterogeneity of 22q11.2 deletion syndrome results in diverse presentations, both in type and severity of difficulties and the age at which they develop, meaning children present to a number of different specialities. Not every child is severely affected, and many will grow up to have a normal life expectancy (1, 6, 8). Some of the core features are shown in the diagram below.
Other useful contacts
Max Appeal is a registered charity, offering support to those affected by 22q11 Deletion Syndrome, their families, carers and friends. They can offer a listening ear, useful literature and help in finding further information. Where possible, a representative of the charity will also be available on the day of the MDT clinic.
Contact Details: www.maxappeal.org.uk
Investigations to consider ahead of referral to facilitate assessment:
Please see referral information document above.
Immunisation:
- Fully immunize promptly except for live vaccines.
Importance of local support services
- Prompt referral to the Paediatric Community Services for assessment and follow up is essential.
- Involvement of therapists for physiotherapy, occupational and speech therapy varies according to individual case.
Annual monitoring including:
- Full blood count for cytopenias, serum calcium and thyroid function
- Height and weight
- Monitor for autoimmune disease, with autoantibody testing as clinically indicated
- Regular dental care
Mental health:
There is increasing recognition of the importance of mental health. Consider Child and Adolescent Mental Health Services referral for assessment if autistic-spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD) and behavioural issues in the preschool and school age child cause dysfunction.
References
1. McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, et al. 22q11.2 deletion syndrome. Nat Rev Dis Primers. 2015;1:15071. doi:10.1038/nrdp.2015.71
2. Max Appeal. A Bit of Background History. [accessed 25 Mar 2018]. Available from: http://www.maxappeal.org.uk/knowledge/a_bit_of_background_history
3. The International 22q11.2 Foundation Incorporated. 22q11.2 Deletion. 2018 [accessed 24 Mar 2018]. Available from: http://www.22q.org/about-22q/faqs/22q11-2-deletion/#1
4. Bassett A, McDonald-McGinn D, Devriendt K, Digilio M, Goldenberg P, Habel A, et al. Practical Guidelines for Managing Patients with 22q11.2 Deletion Syndrome. J Pediatr. 2011; 159(2):332-339. doi:10.1016/j.peds.2011.02.039
5. Wentzel C, Fernström M, Ohrner Y, Annerén G, Thuresson AC. Clinical variability of the 22q11.2 duplication syndrome. Eur J Med Genet. 2008;51(6):501-510. doi:10.1016/J.ejmg.2008.07.005
6. Habel A, Herriot R, Kumararatne D, Allgrove J, Baker K, Baxendale H, et al. Towards a safety net for management of 22q11.2 deletion syndrome: guidelines for our times. Eur J Pediatr. 2014;173(6):757-765. doi:10.1007/s00431-013-2240-z
7. Fung WL, Butcher NJ, Costain G, Andrade DM, Boot E, Chow EWC, et al. Practical guidelines for managing adults with 22q11.2 deletion syndrome. Genet Med. 2015;17(8):599-609. doi:10.1038/gim.2014.175
8. Oskarsdóttir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population-based study in Western Sweden. Arch Dis Child. 2004;89(2):148-151
Rydym yn croesawu galwadau ffôn yn Gymraeg, Saesneg a Iaith Arwyddion Prydain (BSL) via SignVideo.
We welcome phone calls in Welsh, English and British Sign Language (BSL) via SignVideo.